

The second edition of “Professor’s Corner” included a discussion about the fundamental physical property differences between low molecular mass materials and polymers1. These included differences in flexibility, melting point and glass transition temperature (Tg), as well as the rheological properties of liquids or solutions. In this edition, we will take a closer look at structure/property relationships for polymers. We also will introduce terminology clarification: a recurring topic designed to improve communication among those engaged in UV/EB research and development activities.
The rubber band – A valuable learning aid
The properties of any material depend on its composition and molecular structure. Most rubber bands are made from natural sources of rubber because of the elasticity of such materials2. But, it is the molecular structure of a rubber band – a lightly cross-linked polymer – and the resultant fascinating physical properties it produces that are the subject of this discussion.
Thermodynamics of stretching. When you stretch a rubber band, what do you see? Feel? Observe? Stretch it and immediately touch it to your forehead or chin. What do you feel? Now, with it stretched, release it and allow it to retract back, and then quickly touch it again. What do you feel this time? What would happen if you stretched the rubber band and, while stretched, you warmed it with a hair dryer? Would it expand farther, or would it contract?
Enthalpy Change. When you stretched the rubber band, you should have noted that it was warmer than before stretching. This indicates that the enthalpy change, ∆H, is exothermic (more negative) during stretching. When you then allowed a stretched rubber band to contract, you should have felt that it was cooler to the touch. The process of contraction or relaxation from a stretched position is endothermic; the rubber band is absorbing energy from its surroundings. Note that this is a purely physical process. No change in composition has occurred, yet there is a detectable and reproducible thermodynamic result. Since the composition remained the same before and after stretching, it is clear a structural change at the molecular level has taken place.
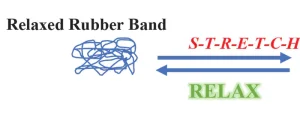
Figures 1 and 2 schematically depict the molecular structure of the rubber band before and after stretching. In the relaxed state, the lightly cross-linked macromolecules are randomly oriented in a purely amorphous form. But, when stretched, they become highly ordered and aligned with one another; they have a high degree of crystallinity when stretched. As the rubber band is elongated, the free volume decreases, and the macromolecules come closer together. This increases the points where they contact one another, causing stronger total forces of attraction. Since all attractive forces are exothermic, thermal energy is emitted during stretching.

Entropy Change. What about the change in entropy, ∆S? Clearly, as the rubber band is stretched, the macromolecules become much more ordered. Therefore, there is a substantial decrease in entropy (∆S = negative). This does not favor spontaneous change and, of course, stretching is not a spontaneous process. Rather, contraction of the rubber band takes place spontaneously when ∆S = positive.
Free Energy Change. While positive changes in entropy favor spontaneity, it is the free energy change of a process, ∆G, that actually determines whether or not a process is spontaneous. To evaluate ∆G, the change in enthalpy, the change in entropy and the Kelvin temperature must all be taken into consideration. Equation 1 shows the relationship among these four parameters.
∆G = ∆H – T∆S Equation 1
When the sign of ∆G is negative, the process will be thermodynamically spontaneous. When the rubber band is stretched, ∆H is quite negative. But, so is ∆S! The entropy change is sufficiently negative when stretched to produce a “T∆S” value more negative than ∆H, and this produces a positive value for ∆G. Thus, stretching is non-spontaneous, while relaxation is spontaneous – even though the former is exothermic and the latter is endothermic. The relaxation process is entropy driven!
So, what happens when you warm a stretched rubber band? If it expands, as many might expect, the temperature (T) would increase and its entropy would decrease. Thus, “T∆S” would become more negative. ∆G would then become more positive. But, if it contracts on heating, T would increase but ∆S also would increase, making ∆G more negative. Thus, a stretched rubber band, when heated, will contract.
These simple thermodynamic observations give good insight into the nature of the rubber band’s macromolecular structure. But, other readily apparent physical properties of a rubber band provide even more insight.
Flexibility, density, modulus and tensile strength. The rubber band is obviously very flexible. But, what happens to its width and thickness as it is stretched? Try it! Since the total volume of the sample will not change, a large change in the length will necessitate a corresponding decrease in the other two dimensions. This provides a mental picture of the macromolecules pulling closer together as they elongate in one dimension. In this process, the free volume decreases and, since the mass does not change, the density increases.
Now try this: Exert a lateral pressure on the relaxed rubber band. What happens? It yields very easily! It has virtually no stiffness, and we can say that its modulus is essentially zero. Now, as you slowly stretch the rubber band, what happens to the modulus? It continues to increase more and more until finally, just before the band breaks, the modulus is at a maximum. Also, as it’s stretched, the rubber band’s tensile strength – its resistance to stretching – increases, becoming stronger and stronger with increasing length. It is obvious, then, that the modulus and tensile strength of a rubber band are proportional to the amount of stretching. Of course, these phenomena have a structural explanation. As with the exothermic nature of stretching, the macromolecules move closer together, aligning more with one another. The increasing total force of attraction caused by the tight alignment of the macromolecules increases both its modulus and its tensile strength.
Other visible effects. In addition to the effects already discussed, a typical brown rubber band will increase in “whiteness” as it is stretched. This effect may be more muted with brightly colored rubber bands. What is happening? This effect is related to an increase in microcrystallinity with stretching. Were there a complete absence of color-producing components in a rubber band, it would likely be transparent. Such transparency reflects its amorphous nature. Highly crystalline polymers, on the other hand – those with a high degree of molecular alignment – tend to be hazy or opaque in direct proportion to the concentration of microcrystalline regions. So, as a rubber band is stretched, it becomes more crystalline and less transparent due to the alignment of the macromolecules. For many rubber bands, this is reflected in increased whiteness. A similar effect is seen when one flexes a transparent plastic such as poly(methyl methacrylate) (PMMA) or polycarbonate (PC). As it is flexed, the polymer “whitens” or shows a “crazing” pattern. This reflects the increased alignment of the macromolecular chains with repeated stretching of the plastic as it is flexed. However, since the plastic is not nearly as elastic as a rubber band, the “crazing” remains after flexing.
Summary. These observations from simple manipulations of a rubber band (or common transparent plastics) provide insight into the macromolecular structure of polymers, contrasting them with low molecular mass materials.
Terminology clarification
People entering the polymer technical workforce are immediately confronted with a wide range of terms and definitions, many of which are used interchangeably and – of greater concern – incorrectly. While there is no perfect vocabulary for the UV/EB professional, it may be useful to confront this linguistic issue directly.
Perhaps a good place to start is with the words “curable,” “cured” or “curing” – as in “UV-curable,” “energy-cured” or “radiation curing.” When a polymer is “cured,” a linear or branched polymeric species is subjected to a subsequent chemical process wherein the macromolecules are linked together by covalent bonds. The terminology comes from traditional conventional thermal or air curing processes. However, this is not typically what happens with UV or EB “curing.” Rather, in most cases, a mixture of nonpolymeric oligomers and monomers of relatively low molecular mass are coated or printed onto a substrate and, subsequently, they are polymerized and cross-linked (“cured”) simultaneously. While this is not a critical linguistic issue, it is more accurate to suggest the terms “UV polymerization,” “EB polymerization” or “energy polymerization” be used in technical papers and presentations to describe the process. Likewise, in speaking of the raw materials (oligomers and monomers) in a typical formulation, it is suggested that these be referred to as “UV-polymerizable” or “EB-polymerizable” materials. Certainly, these materials are being “cured,” but not in the conventional thermal-cure sense. Rather, they are nonpolymeric materials that, when exposed to UV or EB sources of energy, polymerize and cross-link in situ.
Technical questions?
What are your technical questions about polymer science, photopolymerization or other topics concerning the chemistry and technology of UV/EB polymerization? Please submit your questions or comments to Dianna Brodine, managing editor for UV+EB Technology, at dianna@petersonpublications.com.
References for further study:
“Professor’s Corner,” UV+EB Technology, Volume 5, No. 2, 2019.
https://web.archive.org/web/20061018005348/http://www.madehow.com/Volume-1/Rubber-Band.html
 Byron K. Christmas, Ph.D.
Byron K. Christmas, Ph.D.
Professor of Chemistry, Emeritus
University of Houston-Downtown
b4christmas@gmail.com



