

By Nicole L. K. Thiher, Chemical and Biochemical Engineering Department, University of Iowa; Erin Peters, Rockridge High School; Sage M. Schissel, PCT Ebeam and Integration, LLC; and Julie L. P. Jessop, School of Chemical Engineering, Mississippi State University
Abstract
In this study, a protocol was developed to investigate UV- and EB-polymerized films of equivalent radical concentrations. This protocol then was applied to an acrylate/methacrylate pair to characterize the impact of the initiation mechanism. Raman spectroscopy was used to determine differences in polymer conversion. Monomer chemistry was shown to be a key variable in the comparison of the two initiation mechanisms.
Introduction
Two common techniques for radiation-induced polymerization are ultraviolet (UV) and electron beam (EB). Both techniques have advantages over thermal initiation and can be used to create similar products,1,2 including inks, adhesives and coatings.3 Although UV and EB polymerization techniques are similar in many respects, there are differences between the two initiation mechanisms that can affect the polymerization kinetics and polymer properties.4-7
A major difference in UV and EB initiation is how primary radicals are formed. UV requires a photoinitiator, which absorbs light and decomposes to form predictable radical structures.8,9 In contrast, EB has enough energy to form radicals on any molecule in the formulation – even on the body of a polymer chain – and does not require an initiator.10,11 Radical formation during EB exposure is much less predictable than during UV exposure due to this ability to form radicals nearly anywhere. Non-selectivity can result in secondary reactions, such as crosslinking, instead of the desired secondary initiation reaction that is required for polymer formation.12
Other studies have shown that monomer chemistry is a key factor in radical formation and secondary reactions during EB polymerization.12,13 In one study, an increase in labile bonds in a monomer led to a higher concentration of primary radicals. However, this increase in primary radical concentration did not necessarily lead to an increase in the rate of polymerization or a higher final conversion.12 Furthermore, this study also showed differences when comparing the conversion of acrylates and methacrylates. These differences are consistent with major kinetic differences observed during UV-initiation of acrylates and methacrylates in the past.14-18 These previous studies show there is a difference between the kinetics of acrylate and methacrylate polymerization; however, the authors have not found any studies that compare the impact of initiation mechanism on the kinetics of radiation-induced polymerization of acrylates and methacrylates.
In fact, few studies have focused on comparing the kinetics of UV and EB polymerization in general.5-7 Schissel et al. found differences in conversion and physical properties of polymers initiated with UV and EB using equal energy deposition.19 Comparing UV and EB with equal initiation energy highlighted some of the differences in the initiation methods, but other questions remained unanswered.
It is known that radical formation, concentration and secondary reactions are reliant on monomer chemistry, which affects polymerization kinetics. The comparison of UV and EB polymerization at equal primary radical concentrations will reveal the differences in kinetics and network formation driven by monomer chemistry and initiation mechanism. To compare initiation mechanisms, an acrylate/methacrylate pair was exposed to UV and EB, resulting in equal primary radical concentrations. Using Raman microscopy to measure conversion, a relationship among formulation chemistry, initiation mechanism and polymerization kinetics was elucidated for these acrylate/methacrylate systems.
Experimental
Materials
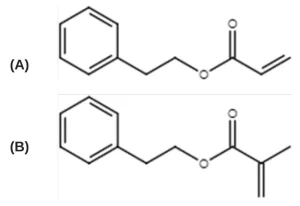
An acrylate/methacrylate pair was chosen to further study the significance of monomer chemistry and initiation mechanism on radiation-induced polymerization kinetics:12,19 2- phenylethyl acrylate (PEA, TCI America) and 2-phenylethylmethacrylate (PEMA, Polysciences) (Figure 1). To induce UV polymerization, the photoinitiator 2,2-dimethoxy-2phenylacetophenone (DMPA, Sartomer) was used. The free-radical inhibitor 2,2,1-diphenyl-1picrylhyrazl (DPPH, TCI America) was used to quantify radical formation. All materials were used as received and stored at room temperature.
Methods
Radical concentration

The method to quantify primary radical concentration during EB exposure has been described in detail previously,20 and only a brief outline of the method is presented here. In previous ionizing radiation literature, the radical concentration was reported in terms of a radiation chemical yield or G-value. Here, radical concentration is reported in mol/L as opposed to radicals per 100 eV or mol/J, as are typically used to report G-values. The unit change was made to accommodate discussion of both EB and UV initiation mechanisms since G-values are associated with EB polymerization but not with UV polymerization.
To determine the primary radical concentration in mol/L, the rate of radical formation (𝑅R) was multiplied by the reaction time. 𝑅R was determined by adding an inhibitor to the monomer formulations, which reacted with primary radicals upon their formation during EB exposure. The disappearance of inhibitor, marked by a color change, was directly proportional to the rate of radical formation (𝑅R) and was monitored using UV-Vis spectroscopy (DU-62 Spectrophotometer, Beckman).
Once the primary radical concentration of each monomer was determined for EB exposure, a program written by Kenning et al. was used to determine the UV requirements necessary to match the radical concentration during UV exposure.21 This program used a set of differential equations to model polychromatic illumination based on initiator concentration, absorbance and efficiency. For each monomer, the concentration of photoinitiator was set at 0.14 mol/L, which was the lowest concentration that allowed the UV radical concentration targets to be achieved with the available lamp. Low initiator concentration was desirable because high concentration of initiator is known to block UV light penetration.8 The exposure time was set at 1 s to match the exposure time of the EB reactions. Matching the exposure time meant that not only would the total radical concentration of the UV and EB reactions be the same, but the average rate of radical formation also would be equivalent. For the program, the quantum yield of DMPA was estimated to be 0.2.22
Conversion measurements

EB sample preparation. Neat monomer was pipetted onto a glass slide, and a tape spacer was used to achieve a sample thickness of ~100 µm. Samples were polymerized using an EBLab unit (Comet Technologies, Inc.) or an EB Pilot Line (BroadBeam EP Series, PCT Ebeam and Integration, LLC). The voltage of both EB units was set at 200 kV to ensure uniform energy deposition throughout each sample. Nitrogen flow was used to reduce the oxygen concentration to less than 200 ppm to minimize the effects of oxygen inhibition. Ten exposure conditions were used for the EBLab experiments, all at a constant dose rate of 197±4 kGy/s. The dose rate was held constant because altering the dose rate has been shown to have an impact on conversion.13,23,24 The dose and line speed combinations match those used to determine the G-values on the EBLab unit and are shown in Table 2. A single 400 kGy exposure at 1.5 m/min was carried out on the EB Pilot line, which was used to gather conversion data rather than determine radical concentration.
UV sample preparation. Monomer containing 0.14 mol/L DMPA was pipetted onto glass slides with a tape spacer used to achieve a sample thickness of ~100 µm. Samples were polymerized using an Omnicure® S1000 Ultraviolet/Visible Spot Cure System (Excelitas, 250-450 nm band pass filter) with a 3-mm liquid light guide at ambient temperature. The tip of the light guide was inserted into a nitrogen-purging chamber. The air gap between the sample surface and the light guide was ~1 mm. Nitrogen flow was used to reduce the oxygen level below 200 ppm to minimize the effects of oxygen inhibition. The effective irradiance was measured by a radiometer (Versaprobe Pro, Con-Trol Cure).
Raman microscopy. Raman microscopy was used to determine conversion of the samples after polymerization. In order to eliminate error from instrumental variation and EB bombardment, a reference peak was used. Previous work has established the reaction peak at 1636 cm-1 (indicative of the -C=C- bond in the (meth)acrylate moiety) and a reference peak at 1613 cm-1 (indicative of the -C=C- bonds in the phenyl ring).25 Fractional conversion, α, was calculated using the following equation:
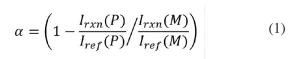
where Irxn(𝑃) and Iref(𝑃) are the peak intensities of the reaction and reference peak of the polymer, respectively; Irxn( (𝑀) and Iref (𝑀) are the peak intensities of the reaction and reference peak of the monomer, respectively.26
Samples were transferred to aluminum Q-panels for analysis. Raman spectra of the samples were collected using an optical microscope (DMLP Leica) connected to a modular research Raman spectrograph (HoloLab 5000R, Kaiser Optical Systems, Inc.) via a 100-µm collection fiber. A single-mode excitation fiber carried an incident beam of 785-nm near-infrared laser to the sample through a 10x objective with a numerical aperture of 0.25 and a working distance of 5.8 mm. Laser power at the samples was ~8 mW. Spectra were collected with an exposure time of 30 s and 3 accumulations. Ten monomer spectra were collected and averaged to provide accurate values for Irxn(𝑀) and Iref (𝑀) to use in Equation 1. The error in the conversion measurements due to instrumental variation is expected to be ±0.05.
Results and Discussion
To compare UV- and EB-initiated polymerizations, an acrylate/methacrylate pair was examined. Polymerization kinetics were studied for UV and EB initiation with equal primary radical concentration. After samples were exposed to either UV or EB radiation, Raman microscopy was used to determine monomer conversion.
Comparison of radical concentration and initiating energy
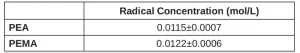
Using the free-radical inhibitor (as described in the Methods section), primary radical concentration during EB exposure was quantified (Table 3). The methacrylate PEMA has three additional labile C-H bonds compared to its otherwise identical acrylate counterpart (PEA) due to the added methyl group. Although these additional labile bonds provide additional locations for primary radical formation, PEA forms approximately the same concentration of radicals (within error) as PEMA.
In order to generate this same number of primary radicals, the energy required for EB is much different than that required for UV. For EB, the monomers received a dose of ~200 J/g. When using UV, the energy was varied to match the radical concentration of the EB samples. The more radicals produced during EB exposure, the higher the effective irradiance required to form the same quantity of radicals. The effective irradiance (I) in was converted to dose (𝐷) in using the exposure time (𝑡), sample mass (𝑚), and area (𝐴) as follows:19
 Note that Equation 2 contains the implicit unit conversions:
Note that Equation 2 contains the implicit unit conversions: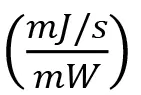 and
and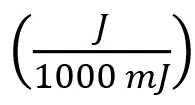 . The EB and UV equivalent doses are compared in Table 4.
. The EB and UV equivalent doses are compared in Table 4.

EB initiation requires ~3 to 4 times the amount of energy as that required by UV to form the same number of primary radicals. The smaller energy requirements for UV initiation are likely the result of differences in bond lability of photoinitiatiors compared to monomers. For example, the photoinitiator DMPA requires 52.1 kcal/mol to generate radicals during UV exposure;27 breaking relatively weak C-H bonds on monomer molecules requires ~100 kcal/mol during EB irradiation.12 Additionally, there are many other reactions that can happen besides radical formation when electrons interact with matter.11 The inefficiency of accelerated electrons is not well established; however, photon loss to other reactions is characterized through the quantum yield of the photoinitiator.
Comparison of monomer conversion
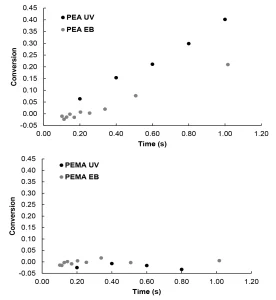
For this study, EB and UV polymerizations were initiated at conditions to produce equal primary radical concentrations in order to compare the impact of initiation mechanisms on radiation-induced polymerization kinetics. The conversion was then measured using Raman microscopy (Figure 2). UV initiation of PEA was faster than EB initiation. Additionally, higher final conversion was observed for UV initiation compared to EB initiation (Figure 2A).
The lower conversion of PEA during EB initiation compared to UV initiation could be the result of differences in primary radical structures that cause differences in secondary radical reactions. UV initiation typically results in predictable radical structures, while EB initiation tends to form many radicals with structures that are much harder to predict.8,10 During EB polymerization, some of the primary radical structures may undergo secondary radical reactions – such as crosslinking, recombination or termination – which do not lead to chain initiation.28 If fewer primary radicals are used to initiate polymerization, conversion is suppressed.
In contrast to PEA, PEMA achieved nearly zero conversion regardless of initiation mechanism (Figure 2B). Previous studies have shown that methacrylates typically react much slower than their acrylate counterparts during UV illumination,14-18 and this trend holds under EB exposure as well. The lack of conversion of PEMA compared to PEA is likely due to the stability of the initiating methacrylate radical compared to the initiating acrylate radical.
Conclusions
The conversion of an acrylate/methacrylate pair was studied for UV and EB polymerization initiated with equal radical concentrations. Higher conversion of the acrylate monomer was achieved with UV initiation when compared with EB initiation. Although both monomers start with equivalent primary radical concentration, EB may be less likely to form initiating radicals compared to UV because of the non-selectivity of the accelerated electrons, which results in less secondary reactions leading to monomer conversion.
For both UV and EB initiation, monomer chemistry has a large impact on polymerization kinetics. Methacrylates had significantly lower conversion than their acrylate counterpart due to the increased stability of the tertiary methacrylate propagating radical compared to the secondary acrylate propagating radical. Knowing the differences that initiation mechanisms and monomer chemistry can make on polymer development can help guide formulation practices and extend the use of radiation-cured materials in industrial processes.
Acknowledgments
This work was supported by the National Science Foundation [grant number 1264622]. The authors would like to acknowledge Stan Howell for his contribution to the development of the nitrogen inerting chamber for UV polymerization.
References
- Cleland, M. R., Parks, L. A., Cheng, S., 2003. “Applications for radiation processing of materials,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 208: 66- 73. doi:10.1016/S0168-583X(03)00655-4.
- Cleland, M. R., 2006. “Industrial applications of electron accelerators.” doi:10.5170/CERN-2006-012.383.
- Cohen, G., 2012. “UV/EB Market Trends,” RadTech Report. 26(2): 44-48.
- Chmielewski, A. G., Haji-Saeid, M., Ahmed, S., 2005. “Progress in radiation processing of polymers,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 236: 44- 54. doi:10.1016/j.nimb.2005.03.247.
- Batten, R. J., Davidson, R. S., Wilkinson, S.A., 1991. “Radiation curing of an epoxy-acrylate-6,7-epoxy-3,7- dimethyloctyl acrylate,” Journal of Photochemistry and Photobiology A: Chemistry, 58: 115-22. doi:10.1016/1010-6030(91)87102-2.
- Patacz, C., Coqueret, X., Decker, C., 2001. “Electron-beam initiated polymerization of acrylate compositions 3: compared reactivity of hexanediol and tripropyleneglycol diacrylates under UV or EB initiation,” Radiation Physics and Chemistry, 62: 403-10. doi:10.1016/S0969-806X(01)00209-2.
- Glauser, T., Johansson, M., Hult, A., 2000. “A comparison of radiation and thermal curing of thick composites,” Macromolecular Materials and Engineering, 274: 25-30. doi:10.1002/(SICI)1439- 2054(20000101)274:1<25::AID-MAME25>3.0.CO;2-L.
- Fouassier, J. P., 1995. “On the structure/properties relationships in photoinitiators of polymerization,” Polymer Engineering & Science, 35: 1061-65. doi:10.1002/pen.760351215.
- Fouassier, J. P., 1995. Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications (Hanser).
- Makuuchi, K., Cheng, S., 2012. Radiation processing of polymer materials and its industrial applications (John Wiley & Sons).
- Richter, K. 2007. Pulsed electron beam curing of polymer coatings (Ph.D. dissertation).
- Thiher, N. L. K., Schissel, S. M., Jessop, J. L. P., submitted 2019. “The Influence of Monomer Chemistry on Radical Formation and Secondary Reactions During Electron-beam Polymerization,” Journal of Polymer Science, in review.
- Chapiro, A., 1962. Radiation chemistry of polymeric systems (Interscience Publishers: New York).
- Takács, E., Wojnárovits, L., 1995. “Comparison of the reactivity of acrylate and methacrylate monomers,”
Radiation Physics and Chemistry, 46: 1007-10. doi:10.1016/0969-806X(95)00310-T.
- Shima, M., Kotera, A., 1963. “Copolymerization characteristics of methyl acrylate and methyl methacrylate. I. Study of copolymerization kinetics by a deuterium tracer method,” J. Polym. Sci. A Gen. Pap., 1: 1115-21. doi:10.1002/pol.1963.100010404.
- Ghorpade, R., Bhosle, S., Ponrathnam, S., Rajan, C., Chavan, N., Harikrishna, R., 2012. “Photopolymerization kinetics of 2-phenylethyl (meth)acrylates studied by photo DSC,” J Polym Res, 19: 1-8. doi:10.1007/s10965-011- 9811-3.
- McCurdy, K. G., Laidler, K. J., 1964. “Rates of Polymerization of Acrylates and Methacrylates in Emulsion Systems,” Canadian Journal of Chemistry, 42: 825-29. doi:10.1139/v64-122.
- Anseth, K. S., Wang, C. M., Bowman. C. N., 1994. “Reaction behavior and kinetic constants for photopolymerizations of multi(meth)acrylate monomers,” Polymer, 35: 3243-50. doi:10.1016/0032- 3861(94)90129-5.
- Schissel, S. M., Jessop, J. L. P., 2019. “Quantitative comparison of photo-and electron-beam polymerizations based on equivalent initiation energy,” Radiation Physics and Chemistry, 157: 72-83. doi:10.1016/j.radphyschem.2018.12.023.
- Thiher, N. L. K., Schissel, S. M., Jessop, J. L. P., 2019. “Counting Radical: Methods to Measure Radiation Yields of Monomers in EB Polymerization,” UV+EB Technology, 5(2): 32-40.
- Kenning, N. S., Ficek, B. A., Hoppe, C. C., Scranton, A. B., 2008. “Spatial and temporal evolution of the photoinitiation rate for thick polymer systems illuminated by polychromatic light: selection of efficient photoinitiators for LED or mercury lamps,” Polymer International, 57: 1134-40. Doi:10.1002/pi.2455.
- Kenning, N. L., 2006. Spatial and temporal evolution of the photoinitiation rate in thick polymer systems (Ph.D. dissertation).
- Siyam, T., 1997. “Gamma-Radiation-Induced Preparation of Polyelectrolytes and Its Use for Treatement of Waste Water,” Handbook of Engineering Polymeric Materials, 9: 119-35.
- Woods, Robert J, and Alexei K Pikaev. 1993. Applied radiation chemistry: Radiation processing (John Wiley & Sons).
- Schissel, S. M., Lapin, S. C., Jessop, J. L. P., 2014. “Internal Reference Validation for EB-Cured Polymer Conversions Measured via Raman Spectroscopy,” RadTech Report, 28(4): 46-50.
- Cai, Y., Jessop, J. L. P., 2006. “Decreased oxygen inhibition in photopolymerized acrylate/epoxide hybrid polymer coatings as demonstrated by Raman spectroscopy,” Polymer, 47: 6560-66. doi:10.1016/j.polymer.2006.07.031.
- Tehfe, M. A., Dumur, F., Graff, B., Morlet-Savary, F., Gigmes, D., Fouassier, J. P., Lalevee, J., 2013. “Design of new Type I and Type II photoinitiators possessing highly coupled pyrene-ketone moieties,” Polymer Chemistry, 4: 2313-24.
- Thiher, N. L. K., Schissel, S. M., Jessop, J. L. P., 2019. “Analysis of methods to determine G-values of monomers polymerized via ionizing radiation,” Radiation Physics and Chemistry, 165: 108394. doi:10.1016/j.radphyschem.2019.108394.


