

In the last two installments of Professor’s Corner, the concept of the glass transition temperature (Tg) was introduced and discussed.1, 2 That discussion continues as we look at the structural and compositional factors that influence the Tg – factors such as polymer backbone rigidity, morphology, cross-link density and pendant side-group length, shape and polarity.
Since the Tg is the temperature at which the onset of long-range segmental motion occurs,3 any structural factor that inhibits the free movement of the polymer chain segments will increase the Tg. For this segmental motion to occur, the atoms along the polymer chain must be able to freely rotate around the bonds connecting them. Thus, a double or triple bond within the backbone will inhibit rotation, raising the Tg. Also, any inherently rigid components within the backbone, or any large or bulky pendant side-group that interacts with the polymer chain, will restrict this free rotation.
Morphology
Recall that the Tg is a property of the amorphous domains only, within a polymer sample.2 Microcrystallites do not exhibit a Tg. However, separate amorphous domains within the sample connect through the microcrystallites if they are present. This causes the amorphous regions to have increased difficulty in achieving the “onset of segmental motion.” So, the higher the microcrystallinity, the higher the Tg of the amorphous domains is expected to be.
Cross-link Density
Essentially, all energy-cured formulations are cross-linked. This cross-linking can preclude alignment of polymer chains, creating more amorphous character. So, how does cross-linking affect the Tg of the amorphous domains? Higher cross-link density polymers have cross-links that are closer together than in lower cross-link density materials. The sites where the chains link up – the cross-links – inhibit the free rotation of the chemical bonds near them. Also, the cross-links themselves may have structures that restrict free rotation. Therefore, the higher the cross-link density, the more inhibition to free rotation, meaning that a higher temperature is needed for the onset of segmental motion. At a sufficiently high cross-link density, segmental motion may be completely inhibited, precluding the effective measurement of the Tg.
Recall from “Part 2” of this Tg discussion that a DMA scan of a cross-linked polymer will show higher storage moduli (“stiffness”) in the “rubbery plateau” at higher cross-link densities. This increase in “stiffness” indicates – along with the value of the peak of the Tan d curve – that the Tg of the polymer has increased.
Pendant Side-Group Effects
The pendant side-groups along the backbone of the polymer result from the selection of monomers and oligomers. Consider polyethylene (PE), polypropylene (PP), poly(1-butene) and longer poly(1-alkene)s. PE has no pendant side-groups. With PP, however, there are methyl groups (CH3-) hanging from the ~[C-C]n~ backbone. And likewise, there are ethyl side-groups (CH3CH2-) along the backbone of poly(1-butene), n-propyl side-groups (CH3CH2CH-) along the backbone of poly(1-pentene), etc., etc. The Tg values of this homologous series of nonpolar hydrocarbon-based polymers provides insight into the effects of side-group length on the polymer Tg.
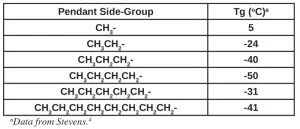
The first effect expected is that the side-groups will prevent strong alignments among the macromolecules. Thus, in general, one might expect the Tg to decrease as the side-group chain length increases. And this is the case, generally speaking. Table 1 indicates that from PP, with its one-carbon side-group, to the four-carbon side-group of poly(1-hexene), this trend is evident. However, for poly(1-heptene), with its five-carbon side-group, this trend is reversed! This indicates that, at this particular length, the side-groups begin to interact more strongly, limiting free rotation. But then, at poly(1-decene), the trend reverses yet again!
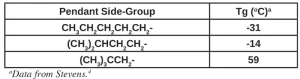
Besides the length, the shape or bulkiness of the side-group also has a predictable effect on the Tg of the polymers. Side-groups, e.g. isopropyl, isobutyl and tertiary-butyl – and isomers of longer alkyl side-groups – do exhibit higher Tgs than their n-alkyl isomers. This indicates a stronger interaction among these bulkier side-groups, further restricting free rotation. Table 2 provides Tg data for the isomers of the n-pentyl side-groups.

The previous examples, of course, are for nonpolar pendant side-groups. It is expected that polar side-groups will cause significantly stronger interactions among macromolecules and their side-groups, and this, in fact, is observed. Comparing the Tg of PP with that of poly(vinyl chloride) (PVC) and poly(vinyl alcohol) (PVOH) illustrates this. While the pendant side-group for PP is nonpolar, the chlorine atom that is pendant to PVC is quite electronegative compared with the carbon atom to which it is bonded, producing a significant dipole-dipole attraction among macromolecules. Then, for PVOH, there is an even stronger macromolecular attractive force: hydrogen-bonding (H-bonding). Table 3 provides Tg data for these three polymers.
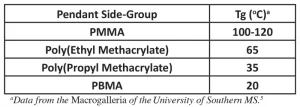
In some cases, the pendant side-groups of polymers may exhibit both length and polar effects. Table 4 provides Tg data for a homologous series of methacrylate monomers from poly(methyl methacrylate) (PMMA) to poly(butyl methacrylate) (PBMA). As the pendant side-group increases in length, just as with the poly(1-alkenes), the Tg decreases. But another factor is at work here. The poly(alkyl methacrylates) all contain a polar carboxyl group (COO-). This makes the side-groups polar. But as the nonpolar alkyl chain gets longer, the overall polarity of the pendant side-groups decreases. So, the strength of the dipole-dipole attractions among the macromolecules decreases. This decreases the resistance to the onset of segmental motion within the polymer, decreasing the Tg. The decrease in both length and polarity, then, results in a lowering of the Tg for these methacrylate polymers.
Summary
The structure and composition of polymers are central factors in the resulting glass transition temperatures. While it may be difficult to determine, a priori, the Tg of a polymer from its unpolymerized components, the basic principles discussed here should allow one to make relative predictions among a related group of polymers.
Technical Questions?
What are your technical questions about polymer science, photopolymerization or other topics concerning the chemistry and technology of UV/EB polymerization? Your question will help guide future topic decisions for this column. Please submit your questions via email to Dianna Brodine at dianna@petersonpublications.com.
References
- Christmas, B. K., “Professor’s Corner,” UV+EB Technology, 6, No. 1, 1st Quarter, 2020, pp. 16-17.
- Christmas, B. K., “Professor’s Corner,” UV+EB Technology, 6, No. 2, 2nd Quarter, 2020, pp. 12-13.
- Stevens, Malcomb P., Polymer Chemistry: An Introduction, 3rd Edition, 1999, pg. 70.
- ibid., Table 3.1, pg. 72.
- https://pslc.ws/macrog/tg.htm, The Macrogalleria, School of Polymer Science and Engineering, University of Southern Mississippi.
 Byron K. Christmas, Ph.D.
Byron K. Christmas, Ph.D.
Professor of Chemistry, Emeritus
University of Houston-Downtown
b4christmas@gmail.com


