

We begin this new year with a continuation of the discussion of microgels, i.e., “nanogels,” introduced in the previous issue of UV+EB Technology.1 Recall from that previous discussion that in UV/EB polymerization processes, particularly those involving free radical addition polymerization of (meth)acrylate-functional monomers and oligomers, a large number of amorphous nanogels are formed within the polymer matrix itself. This creates a very heterogeneous polymer structure at the molecular level. Because of their nanometer -scale size, these nanogels do not typically have a visible effect on the transparency of clear films formed by this process. However, they do cause a significant broadening of the glass transition temperature (Tg), as measured using the tan δ curve of a dynamic mechanical analysis (DMA) scan. They also may have a negative effect on properties such as impact resistance, due to the fact that they represent a different amorphous solid phase within the cured film. Thus, cracks may begin at, and propagate from, the interface between these nanogels and the cross-linked polymer matrix.
How do we know that nanogels form? There are several clues to this. First of all, free radicals and double bonds get trapped within these nanogels2-4 and subsequently, when heated, they react. These post-curing reactions create changes in polymer properties such as modulus, tensile strength, cross-link density, Tg, etc. – properties that are measurable. This reality, by the way, impacts the interpretation of testing results that come from analytical techniques involving heating processes, techniques such as DMA, differential scanning calorimetry (DSC) or differential thermal analysis (DTA). As the polymer matrix expands upon heating and the molecular mobility increases, some number of entrapped radicals and double bonds will react, causing a change in the properties being measured, vis-à-vis those of the unheated sample. This limits the fundamental information that can be obtained about the material properties of the polymer before heating.
To illustrate this, a DMA scan of a UV-polymerized film can be run to characterize the thermomechanical properties of the film. If that exact same sample is then run a second time, without removing it from the sample holder, a different set of properties will, most likely, be observed. The scale of the changes observed will depend on the concentration of entrapped reactive species within the nanogels. In the absence of nanogels, the DMA properties would, most likely, be essentially the same both before and after the second heating.
Further evidence of nanogel formation is given by Kloosterboer.5 He reported that the reactivity of pendant double bonds in growing polymer chains is higher than that of double bonds on the ends of unreacted monomers and oligomers. This higher reactivity leads to a process known as “cyclization” which, then, produces nanogels.
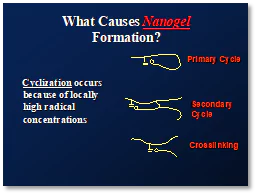
Cyclization. Three different types of cyclization are depicted in Figure 1. Primary cycles form when the growing polymer chain ends react with pendant double bonds along its own backbone. Secondary cycles involve the reaction between pendant side groups on different growing chains. The third type of cyclization produces cross-linking, wherein the terminal double bond of a growing chain reacts with a pendant side group on a different chain. All of these processes lead to the formation of nanogels and to the entrapment of reactive species.
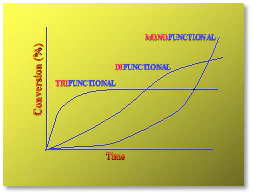
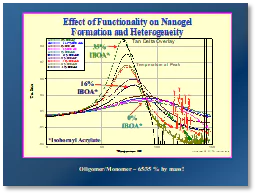
It is well known that higher average functionality (favg) of the monomers and oligomers in UV/EB-curable formulations produces higher overall reactivity and lower percent conversion to polymer (Figure 2). It also is true that higher favg increases the potential for higher concentrations of nanogels to be produced. This is depicted in Figure 3 and was demonstrated in a laboratory experiment conducted in 2014 by students of the Center for Applied Polymer Science Research (CAPSR) at the University of Houston-Downtown (UHD). In that study, 65%-by mass of an acrylate-functional aliphatic urethane oligomer was blended with 35%-by mass of an equal mass mixture of 1,6-hexanedioldiacrylate (HDDA) and trimethylolpropane triacrylate (TMPTA). Isobornyl acrylate (IBOA), a monofunctional monomer, was used in increasing amounts from 0% to 35% by mass as a substitute for the multifunctional monomer blend, reducing the favg of the formulation. Samples then were UV-polymerized and characterized for thermomechanical properties using DMA instrumentation. Results are shown in Figure 4.
From the resulting tan δ curves shown in Figure 4, it readily is apparent that, as the IBOA concentration decreased from 35% to 0% (increasing the favg), the Tg, as measured from the peak of each curve, increased steadily and the width of the curves also increased substantially. The tan δ curve width is directly related to the concentration of nanogels in the film, thus, demonstrating that higher favg produces more nanogels in the cured film. An increase in Tg with increasing favg is fully expected since a higher cross-link density is produced. An increase in nanogel particle concentration is also expected, as the higher functionality of the reactive monomers and oligomers will provide more active sites along the backbone of the growing polymer chains, enhancing the potential for cyclization.
Summary and conclusions. For (meth)acrylate-functional UV/EB-polymerizable formulations, nanogel formation is a fact of life. In most applications, this has not been a problem. But in applications for which it might be a concern, formulators need to be aware of the factors that cause nanogel formation and work to reduce those factors. As with most things, there are trade-offs. Selection of oligomers and monomers with lower overall functionality will reduce the favg, reducing the nanogel concentration. Also, choosing oligomers and monomers with longer “bridge-lengths” – greater distances between double bonds – will help. But both approaches will likely cause a reduction in reactivity and cross-link density, as well as impacting properties such as Tg, tensile strength, modulus, etc. Being fully aware of the issue will allow innovative technical personnel to find creative solutions to this problem.
Technical Questions?
What are your technical questions about polymer science, photopolymerization, or other topics concerning the chemistry and technology of UV/EB polymerization? Your questions will help guide future topic decisions for this column. Submit your questions via email to Dianna Brodine at dianna@petersonpublications.com.
 Byron K. Christmas, Ph.D.
Byron K. Christmas, Ph.D.
Professor of Chemistry, Emeritus
University of Houston-Downtown
b4christmas@gmail.com
References
- Christmas, B. K., “Professor’s Corner,” UV+EB Technology, 6, No. 4, 4th Quarter, 2020, pp. 14-15.
- Ehsan Zarshenas, Saeed Bastani, and Malihe Pishvaei, Ind. Eng. Chem. Res. 2013, 52, pg. 16110.
- Kloosterboer, J. G. Network formation by chain cross-linking photopolymerization and its application in electronics. Adv. Polym. Sci. 1988, 84, 1−61.
- Anseth, K. S.; Anderson, K. J.; Bowman, C. N. Radical concentrations, environments, and reactivities during cross-linking polymerizations. Macromol. Chem. Phys. 1996, 197, 833−848.
- Kloosterboer, J. G.; Lippits, G. J. M. Network formation by photopolymerization: High-precision replication of video discs. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1985, 26, 35-37.



