


In this study, conversion as a function of dose was measured for a series of (meth)acrylates during electron beam (EB) and x-ray polymerization to characterize the impact of the irradiation mode. Monomer chemistry was shown to be a key variable in the comparison of the two technologies, providing guidance for application choices. Part 1 provides the Introduction, Experimental and Methods. Part 2, to be published in Issue 3, 2026, will continue with Results and Discussion.
Introduction
Although x-ray irradiation most often is associated with imaging (medical, security and/or manufacturing inspection), it also holds the potential to be a powerful tool for industrial polymerization and polymer modification applications. X-ray, just as UV or visible light, is a form of electromagnetic radiation, and thus, many of the same benefits that draw users to these other types of radiation polymerization – sustainability, temporal control, mild reaction conditions, etc. – also can be applied to x-ray technology. 1,2 Moreover, x-rays, like the accelerated electrons of EB, are ionizing radiation. Ionizing radiation has sufficient energy to break molecular bonds, making it capable of both excitation and ionization, whereas non-ionizing radiation is limited to interacting with materials through excitation. Because x-ray and EB share this category, both technologies operate similarly on a kinetic and mechanistic level – both do not require an initiator for free-radical reactions; both are colorblind and affected by mass density; and both can be utilized for crosslinking, chain scission and grafting, in addition to polymerization.
However, the use of x-ray, in comparison to EB, introduces an important tradeoff – throughput for penetration. X-rays are generated by converting accelerated electrons using a high Z-value target. This conversion has a low efficiency ranging from ~12% at 7.5 MeV to single percentages for keV electrons. 3 The low conversion between EB and x-ray translates to a significant decrease in the throughput; a dose that a beam can deliver in milliseconds takes an x-ray system minutes to hours to produce. Conversely, without mass or charge, x-ray photons can travel much further than fast electrons of the same energy. X-rays that can penetrate several inches through unit-density material are produced from accelerated electrons that only can penetrate a few hundred micrometers.
This tradeoff is a crucial factor in determining for which applications x-ray is best suited. The penetration ability of x-rays extends the possibilities of utilizing radiation curing well outside the realm of thin films and coatings. Yet, the meager throughput (in comparison to UV and low-energy [≤ 300 kV] EB production) means x-ray will not be cost effective for all processes. The ideal combination for x-ray polymerization or polymer modification is the production of parts that are high-value but low-volume. Examples include curing of composites, post-curing and/or crosslinking of 3D-printed parts, and crosslinking of complex geometries for sensitive applications, such as medical implants. 4-6
In addition to its influence on relevant applications, the variance in throughput between x-ray and low-energy EB also may influence polymer network formation and resultant polymer properties. The throughput of each technology is a result of the dose rate; x-ray systems produce < 1 kGy/s, whereas low-energy EB systems can produce up to 2,000 kGy/s. 1 A slower dose rate means a slower rate of primary radical formation and potentially less radical-to-radical termination. The same total dose may not be required for x-ray-initiated polymerizations as EB, which could reduce exposure time. A slower dose rate also means a longer exposure time to reach a particular dose, which may benefit certain chemistries. Methacrylates, for instance, typically are poorer performers than their acrylate counterparts when EB polymerized, likely due to their radical stability and, therefore, reduced reactivity. 7 A longer exposure time could better align with the propagation rate. Even chemistries that do not exhibit significant dose rate effects over the regime of low-energy EB (i.e., ~10 to 2,000 kGy/s) may experience changes.
In this study, an initial comparison of EB- and x-ray-initiated polymerizations was conducted to ascertain differences between resulting polymers. An acrylate and its analogous methacrylate were chosen to probe the question of reactivity and extended exposure time. Raman spectroscopy was used to determine polymer conversion. Sol-gel analysis was performed to compare network formation.
Experimental
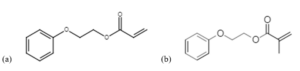
Materials
An acrylate monomer, 2-phenoxyethyl acrylate (POEA, Aldrich), and its analogous methacrylate monomer, 2-phenoxyethyl methacrylate (POEMA, TCI America), were included in the study for comparison to previous EB studies of free-radical polymerization. 7 Consistent with previous studies, the monomers were not washed of inhibitor because the impact of inhibitor was determined to be below the error of the measurements in this study. All materials were used as received (Figure 1).
Methods
EB polymerization
Neat monomer was pipetted onto a glass slide, and a tape spacer was used to achieve a sample thickness of ~100 μm. EB polymerization took place on an EBLab 200 unit (Comet Technologies, Inc.). The voltage was set at 200 kV to ensure uniform energy deposition through the entire depth of each sample. Nitrogen flow was used to reduce the oxygen concentration to less than 200 ppm to minimize the effect of oxygen inhibition.
Ten exposure conditions were used to build the conversion vs. time profile, all at a constant dose rate of ~200 kGy/s. The dose and line speed combinations are shown in Table 1 of Reference 7. Each data point in the profile is a unique sample to avoid any possible effects related to repeated exposure (i.e., pulsed EB). Samples for sol-gel measurements were produced with a dose of 200 kGy at a line speed of 3 m/min.
X-ray polymerization
For conversion vs. time profiles, approximately 0.5 mL of monomer was pipetted into an 11-mm diameter aluminum dish. Once prepared, samples were placed into the x-ray system. A nitrogen purge of 99.999% purity was activated to inert the reaction at a flow rate of 14 SCFM. The purge was run for five minutes prior to the beginning of irradiation and was continued throughout the entire irradiation process. For sol-gel measurements, nitrogen inertization was not available, so samples were prepared by pipetting 0.2 mL of monomer into silanized (Rain-X® treated) Eppendorf tubes. The tubes were capped and further sealed with Parafilm® M to limit oxygen exposure. After irradiation, the tube was cut away for analysis.
Both sets of samples were placed in the x-ray system approximately 5 inches below the x-ray target (eXede, PCT Ebeam and Integration, LLC). The x-ray system was operated at full power, 500 mA and 300 kV, producing a dose rate of ~400 Gy/min (or ~0.007 kGy/s). The accelerating voltage of 300 kV was chosen to maximize the dose rate, since the electron-to-photon conversion is greater at higher voltages. At this voltage, penetration of the x-rays far exceeded the sample thickness, providing a uniform energy distribution. The irradiation time was adjusted for the conversion vs. time samples to change the final dose and build the conversion profile. As with the EB samples, each data point is a separate sample. Samples for sol-gel measurements were irradiated for 75 minutes, equivalent to a dose of approximately 30 kGy.
Raman spectroscopy
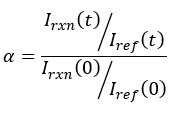 Raman spectroscopy was used to measure the conversion of each sample. Previous work has established, for phenyl-containing (meth)acrylates, a reaction peak at 1636 cm1, indicative of the C=C bond in the (meth)acrylate moiety, and a reference peak at 1613 cm1, indicative of the C=C bonds in the phenyl ring. 8 The fractional conversion then was computed using Equation 1 where I(t) denotes the peak intensity at time t, and I(0) represents the initial peak intensity before irradiation. The subscripts denote whether the measurement is of the reaction peak intensity (rxn) or the reference peak intensity (ref).
Raman spectroscopy was used to measure the conversion of each sample. Previous work has established, for phenyl-containing (meth)acrylates, a reaction peak at 1636 cm1, indicative of the C=C bond in the (meth)acrylate moiety, and a reference peak at 1613 cm1, indicative of the C=C bonds in the phenyl ring. 8 The fractional conversion then was computed using Equation 1 where I(t) denotes the peak intensity at time t, and I(0) represents the initial peak intensity before irradiation. The subscripts denote whether the measurement is of the reaction peak intensity (rxn) or the reference peak intensity (ref).
EB-exposed samples were transferred to aluminum Q-panels for analysis. Raman spectra of the samples were collected using an optical microscope (DMLP Leica) connected to a modular research Raman spectrograph (HoloLab 5000R, Kaiser Optical Systems, Inc.) via a 100-μm collection fiber. A single-mode excitation fiber carried an incident beam of 785-nm near-infrared laser to the sample through a 10× objective with a numerical aperture of 0.25 and a working distance of 5.8 mm. Laser power at the samples was ~8 mW. Spectra were collected with an exposure time of 30 seconds and three accumulations. Ten monomer spectra were collected and averaged to provide accurate values to use in Equation 1. The error in the conversion measurements due to instrumental variation is expected to be ±0.05.
For x-ray-initiated samples, spectra were collected using a handheld Raman spectrometer (BRAVO, Bruker), which utilizes a combination of 785-nm and 852-nm lasers in a method called sequentially shifted excitation (SSE™). Exposure time was variable due to the automated time optimization performed by the instrument software. Conversion vs. time samples were measured using a converted vial tip with a working distance of 3 cm. An average of 30 spectra was taken for the monomers to provide a reference monomer scan. An average of 3 spectra was taken of the irradiated samples, and each condition was replicated in triplicate. The standard deviation for the majority of the samples was ≤ 0.06, although there were some exceptions, especially at short exposure times.
Sol-gel measurements
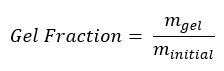 After sample preparation, samples were weighed to record their initial mass, minitial. Next, samples were submerged in 10 mL of tetrahydrofuran (THF). Samples were left in the solvent for a period of at least 72 hours to ensure the linear chains (sol) were removed from the crosslinked structure (gel). Samples then were air dried for six days before being heated at 100° C for 30 minutes to remove the remaining solvent. Once all solvent had been removed, the samples were allowed to return to room temperature and weighed to record their final mass, mgel. The gel fraction for each sample was calculated using Equation 2.
After sample preparation, samples were weighed to record their initial mass, minitial. Next, samples were submerged in 10 mL of tetrahydrofuran (THF). Samples were left in the solvent for a period of at least 72 hours to ensure the linear chains (sol) were removed from the crosslinked structure (gel). Samples then were air dried for six days before being heated at 100° C for 30 minutes to remove the remaining solvent. Once all solvent had been removed, the samples were allowed to return to room temperature and weighed to record their final mass, mgel. The gel fraction for each sample was calculated using Equation 2.
Measurements were replicated in triplicate, and the standard deviation was ≤ 0.07 for all.
Acknowledgements
This work was supported by the National Science Foundation [grant number 1264622].
 Sage Schissel, Ph.D.
Sage Schissel, Ph.D.
Applications Specialist
PCT Ebeam and Integration LLC
sage.schissel@pctebi.com
References
- Spinks, J.W.T., Woods, R.J., An Introduction to Radiation Chemistry, 3rd, John-Wiley and Sons, Inc.: New Jersey, 1990.
- Chapiro, A., 1962. Radiation Chemistry of Polymeric Systems. John Wiley & Sons, Inc., New York, 1962.
- Cleland, M.R., Stichelbaut, F., “Radiation processing with high-energy x-rays,” Radiation Physics and Chemistry, 84, 2013, pp. 91-99. https://doi.org/10.1016/j.radphyschem.2012.06.038.
- Herer, A., Galloway, R.A., Cleland, M.R., Berejka, A.J., Montoney, D., Dispenza, D., Driscoll, M., “X-ray-cured carbon–fiber composites for vehicle use,” Radiation Physics and Chemistry, 78(7–8), 2009, pp.531-534. https://doi.org/10.1016/j.radphyschem.2009.03.047.
- Stevens, L., “X-ray cured carbon fiber composites,” UV+EB Technology, (2), 2022. https://uvebtech.com/articles/2022/x-ray-cured-carbon-fiber-composites/
- Mulliez, M.A., Schilling, C., Grupp, T.M., “Equivalent mechanical properties of X-ray and E-beam cross-linked vitamin E blended ultrahigh molecular weight polyethylene,” Journal of Biomedical Materials Research, 108(5), 2020, pp. 2131-2140. https://doi.org/10.1002/jbm.b.34552.
- Thiher, N.L.K., Schissel, S.M., Jessop, J.L.P., “The Influence of Monomer Chemistry on Radical Formation and Secondary Reactions During Electron-beam Polymerization,” Journal of Polymer Science, 58(7), 2020, pp. 1011-1021.
https://doi.org/10.1002/pol.20190113



