


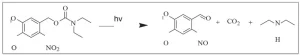
By J. Taylor Goodrich, Alexander Y. Polykarpov and Anushree Deshpande, Sirrus, Inc.
Introduction
It could be said that the modern era of commercial photopolymerization began in 1964 with the installation of the first electron beam cure unit at Boise Cascade for wood finishing. Fifty-five years later, the field of photopolymerization continues to expand: A simple search on Google Scholar for “photopolymerization” from 2019 returns more than 8,440 entries. Acrylates and methacrylates continue to be the materials of choice for the majority of photopolymerization applications, with relatively less attention being spent on epoxides, oxiranes, vinyl ethers/esters and vinyl compounds for reaction with thiols.1 Methylene malonates have been relatively unexplored. The interest in this class of compounds was low, likely due to the lack of commercially viable synthetic routes. Recent developments in the industrial-scale synthesis of methylene malonates2 promise to make new monomers and oligomers based on this chemistry widely available within a few years. With their unique reactivity,3 methylene malonates are likely to make an impact in many polymer applications, including photopolymerization.
Methylene malonates: Unique possibility for dual cure
Methylene malonates contain a carbon-carbon double bond that is 1,1-disubstituted with ester groups. Selected examples of methylene malonate structures are shown in Figure 1. The two ester groups in methylene malonates activate the double bond, making it more electron deficient and more suitable for reaction with electron donors. This activation is weaker than that in cyanoacrylates and is stronger than in acrylates that have only one electron withdrawing group connected to the double bond. Because of this, methylene malonates possess an intermediate reactivity: They do not instantly polymerize on contact with water like cyanoacrylates, yet they can react with weak bases and nucleophiles much more readily and under milder conditions than acrylates and methacrylates. Methylene malonates still can undergo rapid anionic polymerization at room temperature when initiated with a base or a nucleophile.3 Additionally, the carbon-carbon double bond of methylene malonates readily participates in Michael-addition reactions with amines and in catalyzed Michael-addition reactions with polyols.4 The geminal ester groups also can be transesterified while preserving the double bond to create a variety of new monomer structures or, when a polyol is used, a variety of multifunctional methylene malonates. Methylene malonates also can be polymerized via a free-radical mechanism analogous to acrylates and methacrylates. This combination of dual reactivity toward free radicals and nucleophiles coupled with the relative ease of transesterification and low odor of the compounds should allow methylene malonates to become a unique and versatile platform for UV cure applications.
Materials and methods
Materials
Methylene malonate monomers including dihexyl methylene malonate (DHMM), dicyclohexyl methylene malonate (DCHMM) and methylene malonate oligomer based on 1,4-butanediol (BD-PES) were available from Sirrus, Inc. 1,4-butanediol diacrylate (BDDA), 1,4-butanediol dimethacrylate (BDDMA), isobornyl acrylate (IBOA) and isobornyl methacrylate (IBOMA) were obtained from Arkema. Radical photoinitiators 1-hydroxycyclohexyl phenyl ketone and diphenyl(2,4,6- trimethylbenzoyl)phosphine oxide (TPO) were supplied by MilliporeSigma and Rahn AG, respectively. Photolatent base NVOC-DEA was synthesized from commercial products according to Xi, W. et al5. Ethyl 1-methyl-3-piperidinecarboxylate (EMPC) and methanesulfonic acid (MSA) were obtained from MilliporeSigma.
Photopolymerization techniques
Curing profiles were measured by FT-IR during photopolymerization. These samples were cast between glass microscope slides with an 850-micron-thick rubber spacer and irradiated with a mercury arc lamp spot cure system from Synchron, Inc. The irradiance at the sample was 20 mW/cm2 UVA. Free- standing films were prepared by casting liquid photopolymer resin between glass plates with plastic spacers and clamping them together. They were then passed through a Heraeus Noblelight LC6B UV curing conveyor with a Fusion D bulb at a speed of 5 ft/min. Each pass provided 6.7 J/cm2 UVA energy to the sample.
FT-IR
A Perkin-Elmer Spectrum One Fourier-transform infrared spectrometer was used to measure the conversion of carbon-carbon double bonds. For time-resolved measurements the alkene conversion was calculated as the disappearance of the near-IR peak found at 6190 cm-1 in the transmission spectra of the samples. For cured polymer films, the alkene conversion was measured by attenuated total reflectance (ATR) as the fractional decrease in the area of the mid-IR peak occurring at 804 cm-1. These mid-IR spectra were normalized by the area of the carbonyl peak at 1720 cm-1.
DSC
Heat-cool-heat experiments were performed on a TA Instruments DSC 250. Heating and cooling cycles were run at 5°C/min with a maximum temperature of 200°C. The glass transition temperature of samples was calculated from the second heating cycle by the half-height midpoint method.
DMA
Dynamic mechanical analyses were performed to measure tensile properties of prepared polymer films. Tests were performed on a TA instruments Q800 DMA. Multifrequency multistrain tests were run in tensile mode at 1 Hz with 0.1% strain from -60°C to 250°C at a heating rate of 5°C/min. Two heating cycles were recorded for each sample. Glass transition temperatures were recorded as the temperature at which the tan delta peak reached its maximum value. Strain ramp tests were performed at room temperature with a strain rate of 2%/min.
Free-radical cure
Methylene malonates have been thermally, free-radically copolymerized.6 Thermoplastics can be produced with a wide range of glass transition temperatures and degrees of crystallinity. Multifunctional methylene malonates can be incorporated to produce cross-linked materials with excellent solvent resistance as well as high hardness or desired flexibility.
The geminal substitution with two electron withdrawing ester groups leads to resonance stabilization of the radical adducts of methylene malonates and induces steric hindrance in polymerization. Therefore, the propagation of free-radical polymerization of methylene malonates is generally slower than that of acrylates and is more similar to that of methacrylates, where the radical adducts receive additional stabilization from the methyl group, which also increases the steric hindrance.7 Curing speed is important in implementing a photopolymerization chemistry industrially because it saves time, increases productivity and lowers the energy costs of irradiation. It has been recently demonstrated that methylene malonates copolymerize favorably with methacrylates and can enhance the rate of cure of some methacrylates when copolymerized.8
Multifunctional methylene malonates are obtained by transesterification of monomeric methylene malonates with various di- and polyols. Such processes result in mixtures of oligomeric structures, which may enhance the cure due to the additional cross-linking from molecules with higher functionality. In this work, a multifunctional methylene malonate made by transesterification of diethyl malonate and 1,4- butanediol (BD-PES) was compared to 1,4-butanediol diacrylate and dimethacrylate. Monofunctional dicyclohexyl and dihexyl methylene malonates were compared in reactivity with monofunctional (meth)acrylates. This is the first comparison of a multifunctional methylene malonate with a multifunctional acrylate and methacrylate, and the authors strongly believe that more comparisons with different types of oligomers will shortly follow.
Cure profiles
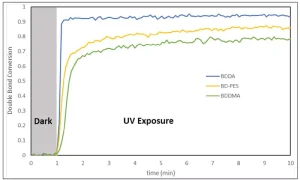
Photopolymer samples were prepared by dissolving 2 wt% 1-hydroxycyclohexyl phenyl ketone photoinitiator in each compound followed by the UV exposure as described in the Materials and Methods section. The specimens were irradiated with 20 mW/cm2 UVA at t = 3 minutes. Double bond conversion was calculated from the area of the near-IR peak found at 6190 cm-1 relative to that peak’s area before irradiation.
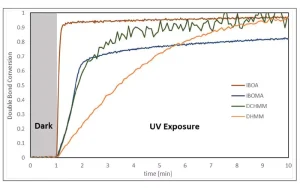
As shown in Figure 2, the room temperature rate of polymerization of the BD-PES methylene malonate was indeed slower than that of BDDA. The initial rate of polymerization of BD-PES was faster than that for BDDMA, and the final double bond conversion was higher. As can be seen from Figure 3, DCHMM cured at a similar rate to IBOMA, while DHMM was slower. Interestingly, while the DHMM cured was slower, it attained a higher double bond conversion than IBOMA and the same conversion as DCHMM and IBOA after 10 minutes of irradiation.
Thermal characterization
Films of UV cured multifunctional methylene malonate and its blends with monofunctional methylene malonates were compared against the cured films of 1,4-butanediol diacrylate. Each of these films was prepared by dissolving 2 wt% 1-hydroxycyclohexyl phenyl ketone photoinitiator in the monomer composition. The compositions were then cast at a thickness of 500 microns and passed through the UV conveyor four times.
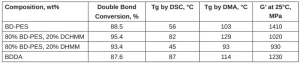
The cured films were characterized for double bond conversion, Tg and tensile modulus. The results can be found in Table 1. The double bond conversion for the BD-PES sample was similar to the BDDA sample, despite the BD-PES sample having an average functionality more than 3 compared to an average functionality of 2 for BDDA. The Tg was 11°C higher by DMA for BDDA over BD-PES. Diluting BD-PES with methylene malonate monomers increased the extent of the double bond conversion in the cured film. Adding 20 wt% of a high-Tg monomer, DCHMM, increased the film Tg by 26°C, and adding 20 wt% of a low Tg monomer, DHMM, decreased the film Tg by 10°C. The storage modulus was 15% higher for BD-PES compared to BDDA.
Photoinduced anionic polymerization
1,4-butanediol-methylene malonate polyester oligomer was mixed with 2 wt% of NVOC-DEA photolatent base (6-nitroveratryl chloroformate coupled with diethylamine). This mixture was shown to be stable (sample was still liquid with no noticeable increase in viscosity) after 14 days.
This mixture was cast between glass plates to obtain a film thickness of 125 microns and then passed through the UV conveyor six times, applying 40 J/cm2 UVA energy to the specimen. The double bond conversion was 38%, and a tacky, free-standing film was produced. The concentration of NVOC- DEA, as well as the applied dose of UV energy, were selected so that the cured film would have sufficient structural integrity but to leave as many methylene malonate double bonds unreacted as possible to maximize the potential for anionic post-cure*. Increasing either the photolatent base concentration or the UVA exposure resulted in a higher double bond conversion immediately after UV exposure. After irradiation, the sample remained at room temperature in the dark for 24 hours. The double bond conversion after 24 hours was 67%. These results indicate that the anionic polymerization of methylene malonate continues after the UV exposure in the dark.

DMA analysis of the photocured film immediately after irradiation and 24 hours later showed an increase in Tg and storage modulus after this post-curing period. The results can be found in Table 2.
2K compositions (UV cure + added base)
Instead of using a photolatent base, anionic post-cure of methylene malonate also can be achieved through the addition of a relatively weak base prior to UV exposure: as long as sufficient pot life is obtained. This allows for the photoinduced free-radical cure to take place before significant anionic cure has occurred.
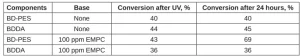
To illustrate this effect, BD-PES and blends of acrylates were prepared with and without the addition of a weak base, ethyl 1-methyl-3-piperidinecarboxylate (EMPC). EMPC was added 5 minutes before a UV exposure of two passes under the UV conveyor system. TPO photoinitiator was used for the free-radical UV cure. The concentration of TPO was 0.2 wt% in the BD-PES samples and 0.01 wt% in the BDDA samples. These concentrations were chosen so that films with approximately 40% conversion of double bonds would be achieved. Limiting the conversion during UV free-radical cure assured the samples would have sufficient level of unreacted methylene malonate groups available for potential anionic post-cure. The double bond conversion of these films was measured immediately after UV irradiation and then again 24 hours later. These results are compiled in Table 3.
The 1,4-butanediol diacrylate was not expected to react with this relatively weak tertiary amine. Therefore, significant post-cure was only observed in the methylene malonate formulation, which was mixed with EMPC before UV irradiation. The BD-PES control test without added EMPC did not post-cure. Thus, the post-cure must have proceeded through the anionic polymerization pathway.
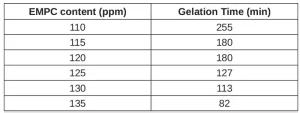
It is important to add the weak base to the methylene malonate photopolymer resin at an appropriate level such that anionic cure is slow enough for the first-stage photoinduced free-radical polymerization to occur before anionic polymerization has significantly increased the viscosity of the resin. Table 4 shows how the pot life of BD-PES can be controlled by varying the level of the weak base added. For these tests, BD-PES sample was first mixed with 50 ppm methanesulfonic acid (MSA) and 2000 ppm TPO photoinitiator (to mimic the presence of photoinitiator in UV cure, though the samples were not exposed to UV this time). EMPC was then dissolved in the samples. Gelation time was recorded as the point at which the samples would no longer flow under their own weight.
Potential for 3D printing
An application that can benefit from anionic post-cure is 3D printing, especially the SLA or DLP types. One challenge faced by UV 3D printing methods is creation of objects with isotropic properties while maintaining high conversion and reducing shrinkage.9 Strength in the direction perpendicular to the cured layers (the z-directional strength) composing a 3D printed object is typically weaker than the strength parallel to the layers. This is due to limited covalent bonding across the printed layers and the tendency of the object to fail adhesively between the layers. As shown in Figure 5, anionic post-cure in a 3D printed methylene malonate system can improve this by creating additional bonds between the printed layers. While UV-initiated radical cure occurs primarily within the layers, anionic cure will occur throughout the entire volume of the object, including between the printed layers.
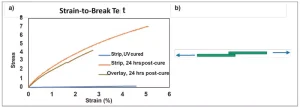
A 500-micron-thick film of BD-PES was UV cured using 0.2 wt% TPO photoinitiator by irradiating with 6 J/cm2 UVA. Before UV curing, 100 ppm of EMPC was added to slowly initiate anionic polymerization. The Young’s Modulus was 0.036 MPa immediately after UV cure, which increased to 2.02 MPa after 24 hours of post-cure time.
To show bonding between layers, a strip of this film was cut and overlaid immediately after UV curing. It was then left to post-cure at room temperature in the dark for 24 hours. The overlaid area was 25% of the total area between the DMA clamps. This overlay sample had a modulus of 1.76 MPa and underwent cohesive failure, showing excellent bonding between the two overlay pieces. If the two overlay pieces were not in contact during the post-cure, they would have individually cured tack-free and showed no adhesion when pressed together.
Conclusion
Methylene malonates were shown to free radically cure at a rate similar to methacrylates. It also was shown that methylene malonates can be UV cured anionically with the use of a photolatent base. UV cure of methylene malonates with a photolatent base led to postcuring of remaining unreacted double bonds in the dark, which was not observed for either methylene malonates or acrylates that were partially polymerized free radically. This anionic post-cure also was observed for methylene malonates when a radical photoinitiator and a weak base were added immediately prior to UV irradiation. Post-cure of double bonds anionically allowed for two partially cured strips of poly-BD-PES to covalently bond together and exhibit significant lap shear strength. Future work will show how these compositions may lead to improved z-directional strength in 3D printed
articles.
References
- Some recent examples of other than (meth)acrylate functional groups used in UV cure can be found in: T. Robert, S. Eschig, T. Biemans, F. Sheifler, “Bio-based polyester itaconates as binder resins for UV-curing offset printing inks,” J. Coat. Technol. Res. 2019, 16, 3, 689-697. G. Peer, A. Eibel, C. Gorsche, Y. Catel, G. Gescheidt, N. Moszner, R. Liska, “Ester- Activated Vinyl Ethers as Chain Transfer Agents in Radical Photopolymerization of Methacrylates,” Macromolecules, 2019, 52, 7, 2691-2700.
- A. Malofsky, T. Dey, J. Sullivan, Y. Chen, S. Wojciak, B. Malofsky, “Synthesis of Methylene Malonates Substantially free of Impurities,” US 8,609,885, 2013.
- B. Malofsky, A. Malofsky, M. Ellison, “Ink Coating Formulations and Polymerizable Systems for Producing the Same,” US 9,234,107, 2016; A. Holzer, A. Deshpande, A. Palsule, J. Sullivan, “Process for UV Curing of Methylene Malonates,” WO 2019014528, 2018.
- A. Palsule, J. Sullivan, K. Vanderpool, A. Holzer, A. Deshpande, J. Klier, “Coatings Containing Polyester Macromers Containing 1,1-Dicarbonyl-Substituted 1-Alkenes,” US 9,567,475, 2017.
- W. Xi, H. Peng, A. Aguirre-Soto, C. Kloxin, J. Stansbury, C. Bowman, “Spatial and Temporal Control of Thiol-Michael Addition via Photocaged Superbase in Photopatterning and Two-Stage Polymer Networks Formation,” Macromolecules, 2014, 47, 6159-6165.
- G. Bachman, H. Tanner, “Preparation of Methylene Dialkyl Malonates,” US 2,212,506, 1939.
- K. McCurdy, K. Laidler, “Rates of Polymerization of Acrylates and Methacrylates in Emulsion Systems,” Can. J. Chem. 1964, 42, 825-829.
- M. Guichard, B. McGrail, W. Wolf, J. Klang, “Curable Compositions and Uses Thereof,” WO 2018219729, 2018.
- A. Bagheri, J. Jin, “Photopolymerization in 3D Printing,” Appl. Polym. Mater. 2019, 1, 593-611.
Acknowledgments
We thank our colleagues Mark Holzer, Aniruddha Palsule and Jeff Sullivan, all of Sirrus, Inc., for helpful discussions and our parent company, Nippon Shokubai, for their support.



