

To understand the nature of solids and liquids, the forces of attraction holding the component atoms, molecules and/or ions together must be considered. Without these forces, only gases would be possible, and the universe would be a very different place. So, for solids and liquids to exist at all, gaseous species must have the inherent ability to attract one another strongly enough to condense into liquids and solids. Relatively low-molecular-mass materials can exist in all three states of matter, but polymers typically exist only as liquids and solids. This fact indicates that the total forces of attraction among polymer macromolecules are very strong indeed. Were one to attempt to evaporate a polymeric liquid or sublime a polymeric solid by lowering the pressure and raising the temperature, decomposition would occur well before any intact macromolecules could break free from the bulk liquid or solid to become independent gaseous species. Also, many solid polymers (including all cross-linked polymers) exhibit sufficiently strong intermolecular attractions that decomposition occurs before they can be melted. A review of these intermolecular forces of attraction is, therefore, in order.
Let us consider three types of intermolecular attractive forces: 1) London dispersion forces, a.k.a., instantaneous dipole/induced dipole attractions; 2) dipole/dipole attractions; 3) hydrogen bonds. These and other types of intermolecular attractive forces also are collectively known as “van der Waals” attractions. In polymer textbooks, they also often are called “secondary-bond forces.”1, 2 It is important to differentiate these attractive forces from the relatively large forces involved in ionic, polar covalent or pure covalent bonds that hold atoms and ions together in molecules or ionic solids. In most cases, individual intermolecular attractive forces are significantly weaker than the chemical bonds found in molecular or ionic species. However, because of their very large size and their ability to entangle with one another, macromolecules in a polymer sample have many sites of contact among the constituent atoms and, thus, the total forces of attraction are very large indeed. In the case of cross-linked polymers, the attractive forces among the macromolecules are, in principle, covalent or polar covalent chemical bonds! This is the reason a cross-linked polymer solid cannot be dissolved or melted. To do either would require the breaking of chemical bonds. But, even in the case of cross-linked polymers, intermolecular attractive forces also are present.
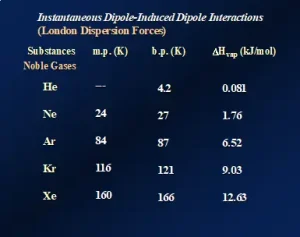
London dispersion forces (LDFs): All atomic, ionic and molecular species, including polymers, exhibit “instantaneous dipole/induced dipole” attractions. This longer name describes the nature of these attractive forces, and their presence is illustrated by the data presented in Figure 1. The data demonstrate the effects of LDFs on three physical properties of the noble gases – Group 18 of the Periodic Table. Going down the group from He to Xe, the melting points, boiling points and heats of vaporization (ΔHvap) all increase, indicating that the forces of attraction among the atoms are increasing.
Going down the group, the atoms get larger, and the number of electrons increases substantially from two electrons for the He atoms to 54 electrons for Xe atoms. The first ionization energy of these elements also decreases with increasing size. The net result of these three changes is an increase in the “polarizability” of the atoms: the ability of their electron clouds to be distorted by the near approach of charged species. The noble gases are, of course, completely nonpolar and monatomic in the gaseous state. So, what is the mechanism of attraction? As the atoms increase in size, one can imagine, in a classical sense, that at any given instant there will be more electrons on one side of the atomic nucleus than on the other. This produces an instantaneous and short-lived charge separation within the atom, i.e., an “instantaneous dipole.” Now suppose that, at the same instant when this dipole forms, another atom contacts it. This second atom’s electron cloud gets distorted, creating an “induced dipole.” This chain of events continues throughout the sample, causing the atoms to attract one another. The higher the polarizability – the higher the ability to distort the electron clouds – the stronger the attractions. Note that He atoms only possess two electrons and are very small. So, they have very low polarizability – so low, in fact, that this element cannot be solidified under normal atmospheric pressure, even when cooled to a fraction of 1K!

Figure 2 provides a schematic illustration of how LDFs function. These forces only work at very short range – the atoms must effectively touch each other while one is in a dipolar state. The spherical model shown in Figure 2, of course, is not a good representation of macromolecules. But due to their enormous size, the constituent atoms and groups of atoms in macromolecules do interact this way, providing for large total forces of attraction. In fact, due to the intramolecular folding of the long polymer chains, LDFs and other secondary-bond forces take place within a given macromolecule as well as intermolecular attractive forces among different macromolecules.
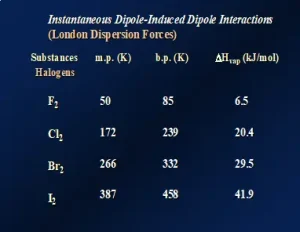
One might think that these data are unique to the noble gases. So, let’s look at a similar series of nonpolar molecular species. Figure 3 illustrates LDFs for the diatomic molecules of the halogens in Group 17 of the Periodic Table. Again, going down this group, the molecules increase both in size and the number of electrons, causing a significant increase in polarizability. The melting points, boiling points and ΔHvap all increase. In fact, in this series, F2 and Cl2 are both gases at room temperature while Br2 is a liquid and I2 is a solid, demonstrating the substantial increase in LDFs as the molecules get larger. Even though it is typical to think of LDFs as relatively “weak” forces compared to dipole-dipole attractions and hydrogen-bonding, LDFs can be quite large when the molecular size and number of electrons are large. This is illustrated by nonpolar I2 having a melting temperature of about 114oC!

It is expected that LDFs will play a significant role in understanding the physical properties of polymers. A key property of linear polymers is the “cohesive energy density” (CED). This can be thought of as the energy required to remove macromolecules from the liquid state per cubic centimeter (J/cm3). Since polymer samples cannot be evaporated, the values for CED must be calculated using methods that are rather complex and are beyond the scope of this discussion. Their primary utility for polymer chemists is to determine solubility parameters that allow for predictions of solubilities of linear or branched polymers in selected solvents3, 4. For our purposes, Figure 4 provides CED data for three well-known nonpolar linear polymers: Polyethylene has no pendent side-groups, polyisobutylene has two aliphatic side-groups and polystyrene has an aromatic side-group. As these macromolecular species increase in complexity and in the monomer’s molecular mass, a higher number of electrons are present and the total LDFs increase, causing an increase in CED.
Most polymers, including those prepared using UV/EB processes, contain bonds that are polar; they have a permanent charge separation. So, in addition to London dispersion forces, these also have dipole/dipole attractions and/or hydrogen-bonding. These secondary-bond forces will be discussed in a later edition of “Professor’s Corner.”
 Byron K. Christmas, Ph.D.
Byron K. Christmas, Ph.D.
Professor of Chemistry, Emeritus
University of Houston-Downtown
b4christmas@gmail.com
References
- Textbook of Polymer Science, Fred W. Billmeyer, Jr., 3rd Edition, John Wiley & Sons, 1984, pp. 12-14.
- Polymer Science and Technology, Robert O. Ebewele, Chapman & Hall/CRC, CRC Press LLC, 2000, pp. 59-63.
- Op. cit., Billmeyer, pp. 151-154.
- Op. cit., Ebewele, pp. 73-76.



